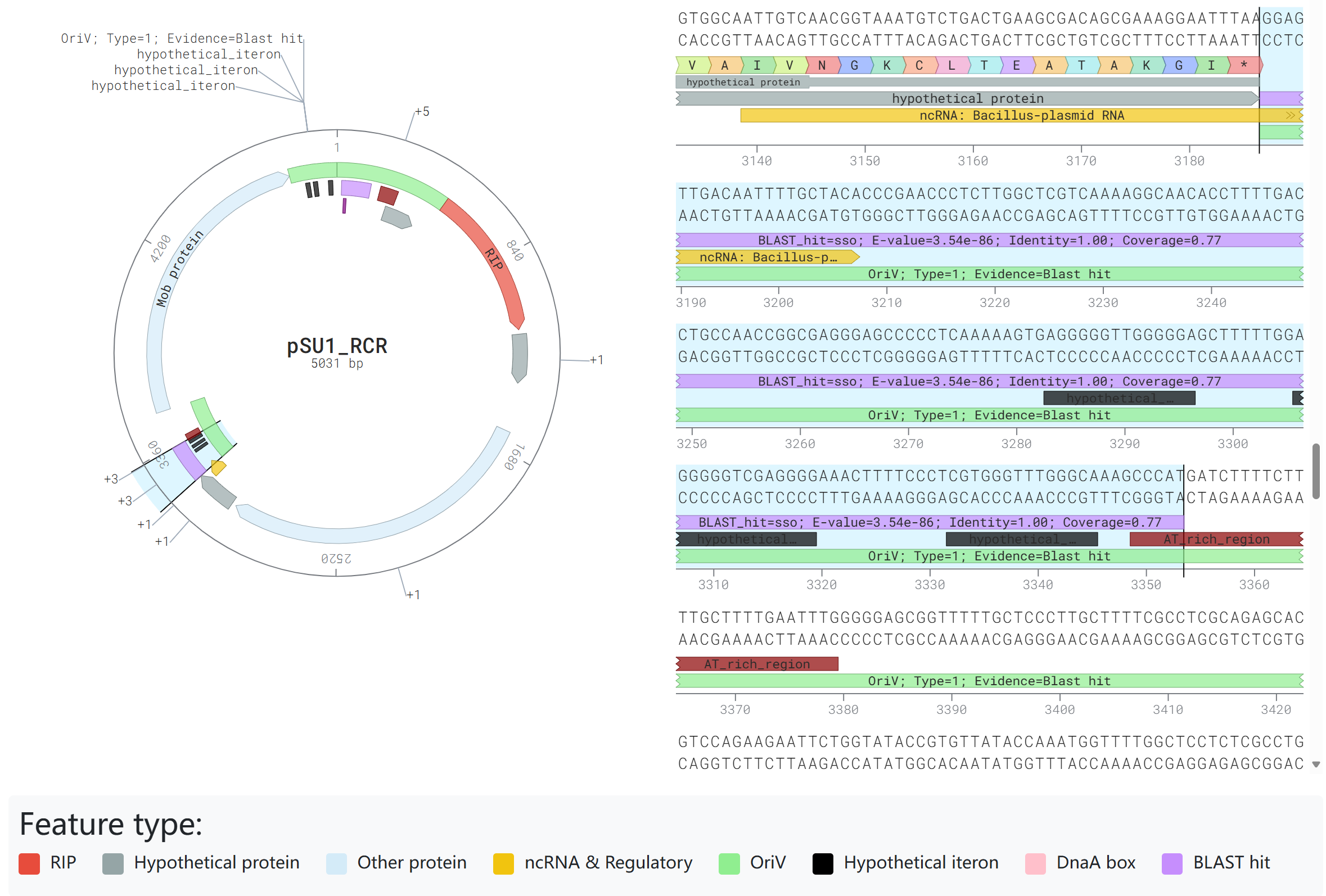
OriV-Finder Analysis Pipeline
To systematically identify the oriVs in plasmids, a bioinformatics analysis pipeline was developed, as shown in the figure below.
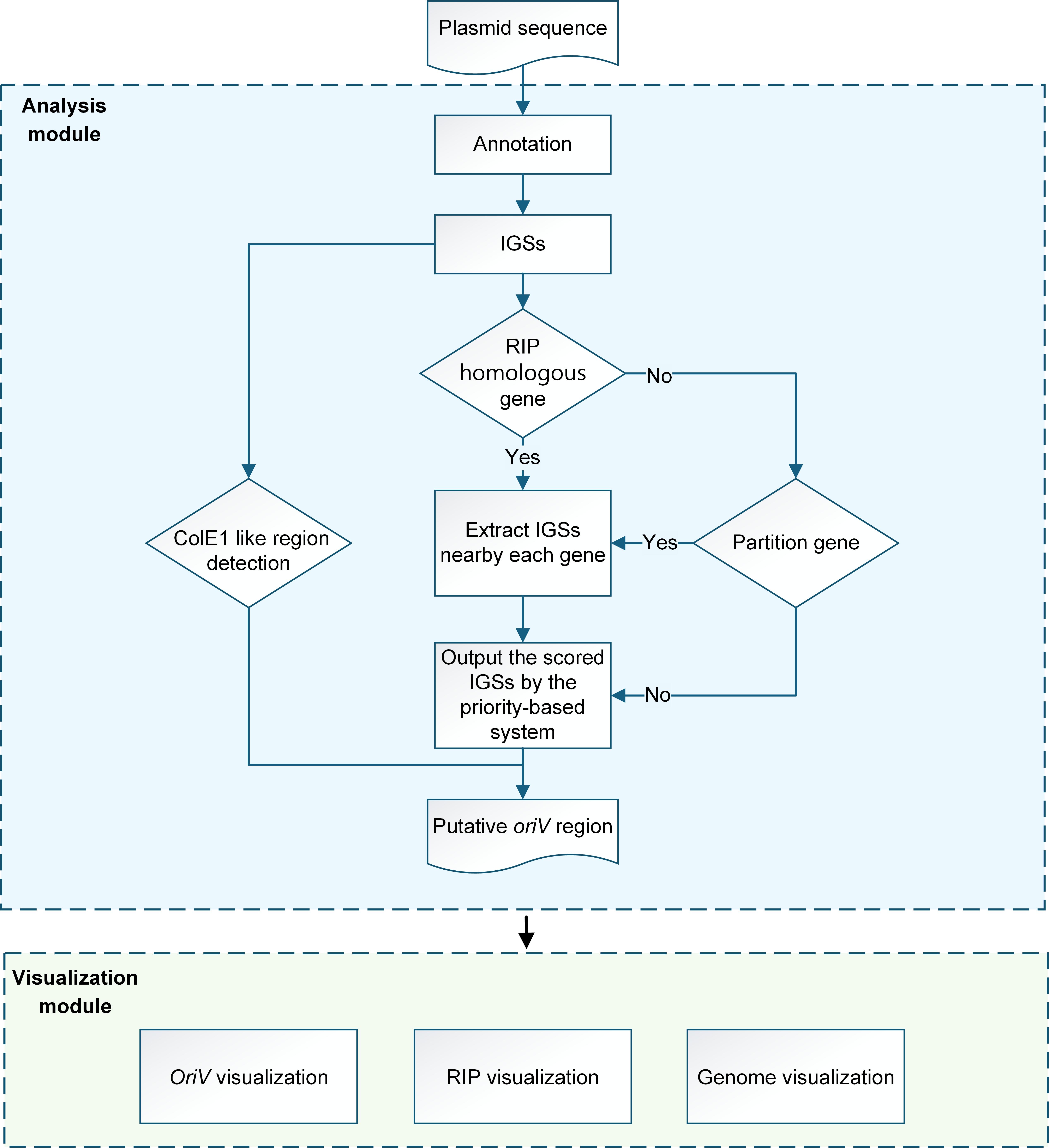
Step 1: The query genome was annotated, and then the CDSs and IGSs were obtained for subsequent processing.
Step 2: Detection of homologous genes of RIPs in CDSs
Step 3: Detection of oriV features in IGSs
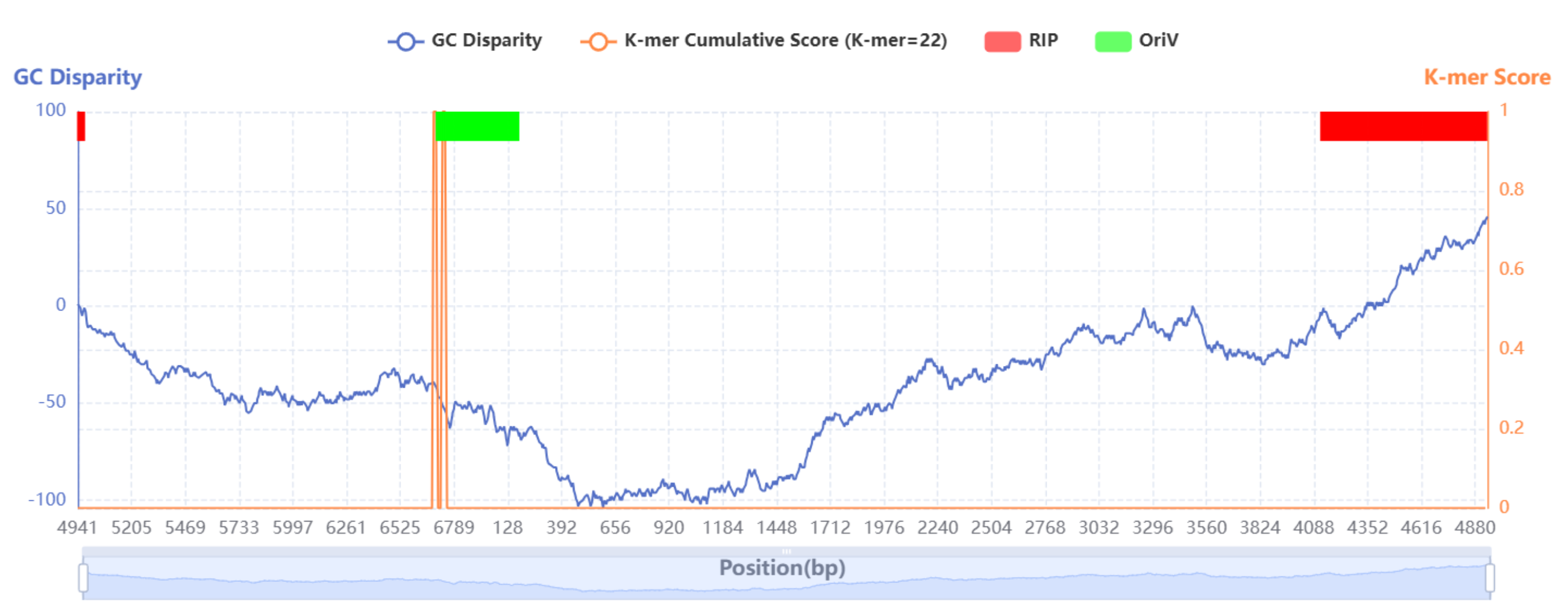
- OriV similar sequences: BLASTN is employed to identify similar sequences to curated oriV regions collected from the DoriC database and literatures;
- ColE1 like regions: The presence of ColE1 like region has been searched for by the Infernal program;
- Iteron sequences: A K-mer sliding window approach combined with Shannon entropy scoring is applied to detect potential iteron sequences;
- AT-rich region: A modified Z-curve-based algorithm is utilized to identify potential AT-rich regions;
- Conserved motifs: Common conserved motifs associated with oriVs are identified.
Step 4: Priority-based scoring system for potential oriVs
Based on the detection results, OriV-Finder employs a priority-based scoring system to identify potential oriVs. Each IGS will be assigned a type by this scoring system, and the IGS with high priority will be output as potential oriVs.
- Type 1 (Highest Priority)
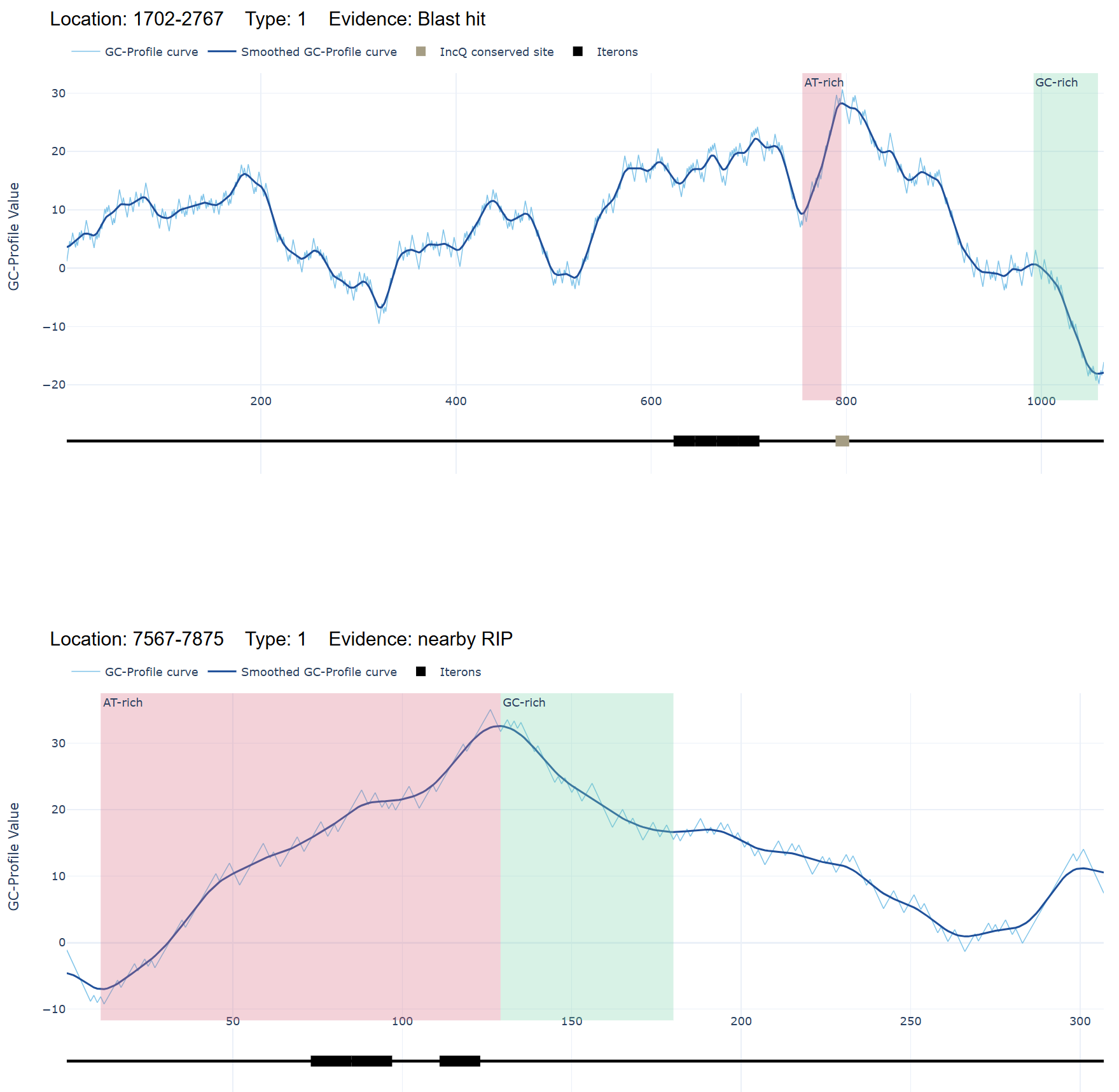
Blast hit presence: If an oriV similar sequence is detected within an IGS, the IGS is designated as Type 1(Evidence: "Blast hit");
ColE1 like region presence: If a ColE1 like region is detected within an IGS, the IGS is designated as Type 1 (Evidence: "ColE1 like");
RIP flanking IGSs: For each identified homologous gene of RIP, the CDS and its four flanking IGSs (two upstream and two downstream) were evaluated, and the region with the highest score among these five segments is designated as Type 1 (Evidence: "RIP" or "nearby RIP"). - Type 2 (Secondary Priority)
No RIP homologs: In the absence of RIP homologs, for each partition protein (like ParA or ParB), the six intergenic sequences (IGSs) flanking the partition protein (three upstream and three downstream) were evaluated. The IGS with the highest comprehensive score among these six candidates is designated as Type 2. *This result should be treated with caution and further analysis is needed. - Type 3 (Lowest Priority)
No RIP or partition protein: If neither RIP homolog nor partition-related protein is present, the IGS with the highest score is designated as Type 3. *This result should be treated with caution and further analysis is needed.